DDI Induction
Cytochrome P450 induction forms part of in vitro experimental ADME services. A new test compound’s induction profile should routinely be evaluated.
These studies include but are not limited to:
CYP1A2, CYP2B6 and CYP3A4
Other Phase I enzymes including monoamine oxidase (MAO), flavin monooxygenase (FMO), xanthine oxidase (XO), and alcohol/aldehyde dehydrogenase
Phase II enzymes including UDP glucuronosyl transferases (UGTs)
Human hepatocytes continue to serve as the system of choice for evaluating enzyme induction in vitro, both with freshly isolated human hepatocytes and cryopreserved hepatocytes available for routine use.
When determining the enzyme induction potential of new test compounds using cultured human hepatocytes or HepaRG™ cells, the following elements must be considered:
- Inter-individual variability
- Changes in mRNA level of target genes as an endpoint (activity level is also evaluated at Eurosafe)
- The inclusion of vehicle controls, positive controls (usually known strong inducers), and negative controls (usually known non-inducers) in the experiment.
Initially CYP1A2, CYP2B6, and CYP3A induction is evaluated (FDA guidance on DDI studies).
If no induction of CYP3A4/5 enzymes is observed, it is unnecessary to evaluate the induction potential of CYP2C enzymes because both CYP3A4/5 and CYP2C enzymes are induced via activation of pregnane X nuclear receptor (PXR).
If CYP3A4/5 induction does occur, however, it is then necessary to evaluate the potential of CYP2C induction.
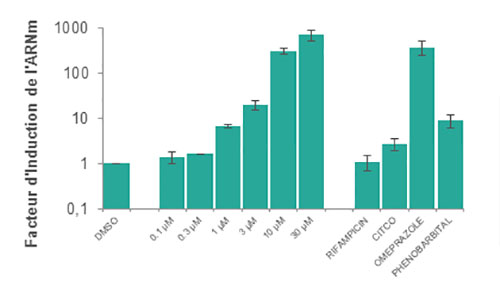
We examine fold-change in CYP enzyme mRNA levels when incubated with the test compound by using cutoffs as described in FDA guidelines.
Eurosafe also evaluates of drug binding to microsomal proteins and cells in order to evaluate free fraction (fumic, fuhep) and also to predict hepatic clearance or drug-drug interaction potential in vivo, utilizing in vitro microsomal metabolic data.
DDI Inhibition
Inhibition of metabolism enzymes (CYP and UGT) by a new test compound may decrease the metabolism of co-medicated drugs. The potential of this test compound to inhibit enzymes is usually investigated to elucidate the precise inhibition mechanisms (reversible or time-dependent) as well as inhibition potency (e.g., Ki). FDA guidelines.
Liver microsomes are recommended for screenings and for mechanistic evaluations. Alternatively for systematic approaches, human hepatocytes or HepaRG™ cells can be used to determine the CYP inhibition potential of test compounds in the cellular environment.
Screening assays are performed with up to 9 cytochrome P450 enzymes (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5) using 10 probe reactions. The Direct Inhibition assay delivers IC50 values towards each CYP enzyme. The Time Dependent Inhibition assay yields direct and shifted IC50 values for each CYP enzyme.
Following IC50 determination, we determine Ki for the test compound against the appropriate enzyme. This parameter elucidates the potency of inhibition as well as the type of inhibition (i.e. competitive, non-competitive, uncompetitive or mixed). Such information can be used to estimate the impact of any potential in vivo interactions.
Eurosafe also evaluates of drug binding to microsomal proteins and cells in order to evaluate free fraction (fumic, fuhep) and also to predict hepatic clearance or drug-drug interaction potential in vivo, utilizing in vitro microsomal metabolic data.
DDI Phenotyping
In order to predict DDI, it is crucial to understand which enzymes (Phase I and phase II) are responsible for the metabolism of a given test compound.
There are three well-characterized methods recognized by the FDA for identifying the individual CYP enzymes involved in a drug’s metabolism.
- Method 1 uses a bank of human liver microsomes characterized for CYP activity
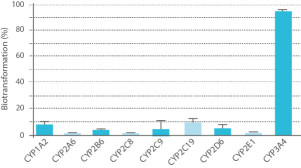
- Method 2 uses individual human recombinant CYP enzymes
- Method 3 uses specific chemicals or antibodies as specific enzyme inhibitors
- Method 3 bis: Eurosafe proposes the use of Silensomes™.
Silensomes™ are validated human-pooled liver microsomes (HLMs) which are chemically and irreversibly inactivated for a specific CYP450 using mechanism based inhibitors (MBI). This approach allows quantitative phenotyping by determining metabolic contribution (fm) instead of RAF evaluation.
- Yannick Parmentier, Corinne Pothier, Audrey Delmas, Fabrice Caradec,Marie-Michèle Trancart, Fabrice Guillet, Belkacem Bouaita, Christophe Chesne, J. Brian Houston & Bernard Walther, "Direct and quantitative evaluation of the humanCYP3A4 contribution (fm) to drug clearance usingthe in vitro SILENSOMES model", Xenobiotica, Volume 47, pages 562-575, 2016.
- Yannick Parmentier, Corinne Pothier, Nicola Hewitt, Ludwig Vincent, FabriceCaradec, Jia Liu, Feifei Lin, Marie-Michèle Trancart, Fabrice Guillet, BelkacemBouaita, Christophe Chesne & Bernard Walther, "Direct and quantitative evaluation of the majorhuman CYP contribution (fmCYP) to drugclearance using the in vitro Silensomes™ model", Xenobiotica, Volume 49, pages 22-35, 2018.
It is recommended that at least two of the three methods above be performed in order to identify the specific enzyme(s) responsible for a given drug’s metabolism.

